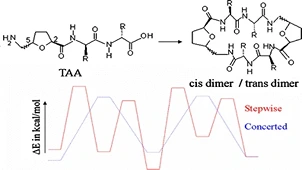
Density functional B3LYP method was used to investigate the preference of intra- and inter-molecular cyclizations of linear tripeptides containing tetrahydrofuran amino acids. Two distinct model pathways were conceived for the cyclization reaction, and all possible transition states and intermediates were located. Analysis of the energetics indicate intermolecular cyclization being favored by both thermodynamic and kinetic control. Geometric and NBO analyses were performed to explain the trends obtained along both the reaction pathways. Conceptual density functional theory-based reactive indices also show that reaction pathways leading to intermolecular cyclization of the tripeptides are relatively more facile compared to intramolecular cyclization.

NVS Kumar, UD Priyakumar, H Singh, S Roy, TK Chakraborty