
GN Sastry, HSP Rao, P Bednarek, UD Priyakumar,
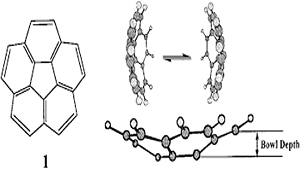
GN Sastry, HSP Rao, P Bednarek, UD Priyakumar,
UD Priyakumar, GN Sastry,
Among the hydrocarbons with (CH)2k (k = 1,2,3,4 …. ) structural formula. (CH)8 where k=4, possesses nineteen local minima at semiempirical level with comparable heats of formation. AM1 procedure is found to be better than MNDO and PM3 in evaluating the heats of formation and geometries based on comparison with available experimental data of the neutral isomers. Vertical and adiabatic electron affinities and ionization potentials have been calculated for all the available isomers. The relative energy ordering is mainly controlled by the electronic factors rather than the strain. Koopman’s theorem is not expected to yield reliable answers for ionization potentials, as the orbital relaxation seems to be very high for some isomers. The major geometric distortions are due to either Jahn-Teller distortions or strong vibronic interactions and the consequent reordering of the skeleton. Relaxation energies from vertical to adiabatic states are roughly proportional to the geometric deformations that occur upon ionization or addition of an electron.
UD Priyakumar, GN Sastry,
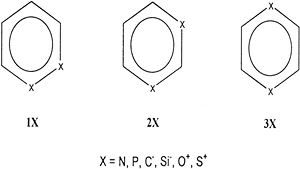
The positional isomers of disubstituted benzenes, (CH)4X2 (X = N, P, C–, Si–, O+, and S+), are studied using ab initio molecular orbital theory at Hartree−Fock (HF), MP2, and CCSD(T) levels and also using density functional theory (B3LYP). All of the planar structures are characterized as minima, and they show fully delocalized geometric parameters, with no significant sign of bond fixation. The ortho isomer is computed to be the least stable in all cases except when X = P and Si–. While meta isomer is more stable than para in general, exceptions are seen for C– and S+ substitutions. Various factors that affect the relative stabilities of positional isomers, viz., (a) lone pair−lone pair (lp−lp) repulsion, (b) electrostatic interaction, (c) bond strengths in the corresponding valence isomers, and (d) topological charge stabilization (TCS), are invoked to explain the computed pattern of relative stabilities. The strengths and limitations of each of these aspects in controlling the geometries and energetics of the three isomeric forms are assessed. The lp−lp interaction is found to be insignificant in the isomers bearing third row atom substituents. The rule of TCS excellently accounts for the distinction between meta and para isomers. The sum of the bond strengths of the constituent Kekule forms derived from bond increment schemes successfully explain the differences in the stability between ortho and meta/para isomers. The effect of skeletal disubstitution on the out-of-plane distortivity in the three isomeric forms is discussed by examining the changes in the frequencies of normal modes. The tendency for ring-puckering increases as more heteroatom substituents from the third row replace the CH groups in the ring. Comparisons were made with the monosubstituted analogues, and the effect of adding one more heteroatom substituent on the out-of-plane distortive modes is analyzed.